
Translate this page into:
Pigmented purpuric dermatosis: A dermoscopic and management perspective


*Corresponding author: Devinder Mohan Thappa, Department of Dermatology and STD, Jawaharlal Institute of Postgraduate Medical Education and Research, Puducherry, India. dmthappa@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Murugan K, Thappa DM. Pigmented purpuric dermatosis: A dermoscopic and management perspective. CosmoDerma. 2024;4:128. doi: 10.25259/CSDM_140_2024
Abstract
Pigmented purpuric dermatosis (PPD) is an acquired chronic relapsing skin condition. It is characterized by asymptomatic or itchy symmetric petechiae, macules, and red-brown patchy pigmentation. The most common age group is between 4th and 5th decades of life. The most common site is the lower limbs. It is divided into 6 types: Progressive pigmentary dermatosis (Schamberg disease, pigmented purpuric lichenoid dermatitis of Gougerot and Blum, purpura annularis telangiectodes (Majocchi purpura), lichen aureus, eczematid-like purpura of Doucas and Kapetanakis, and itching purpura). The dermoscopic features of PPD are well established. They include red dots and globules, coppery red pigmentation, brown patches, brown dots, red patches, linear vessels, annular/comma-like vessels, and lentigines-like reticular pigmentation. Main histopathological features include superficial perivascular lymphocytic and histiocytic infiltration with marked hemosiderin deposition and no leukocytoclastic vasculitis. As there is no standardized treatment, various methods with varying efficacy are employed. This article presents a dermoscopic and management perspective of PPD and reviews the current literature related to this entity.
Keywords
Pigmented purpuric dermatosis
Clinical types
Dermoscopic-histopathology correlation
Treatment options

INTRODUCTION
Pigmented purpuric dermatoses (PPD) include several skin diseases characterized by multiple petechial hemorrhages as a consequence of capillaritis. PPD is an acquired chronic relapsing skin condition. It is characterized by asymptomatic or itchy symmetric petechiae, macules, and red-brown patchy pigmentation.[1,2] The most common age group is between 4th and 5th decades of life.[2] The most common site is the lower limbs.[1]
It is divided into 6 types: Progressive pigmentary dermatosis (Schamberg disease, pigmented purpuric lichenoid dermatitis of Gourgerot and Blum, purpura annularis telengiectoides (Majocchi purpura), lichen aureus, eczematid-like purpura of Doucas and Kapetanakis, and itching purpura.[1,3] Other rare types of PPD that were observed include granulomatous, linear, transitory, quadrantic, and familial forms.[4]
Schamberg disease is the most common type characterized by red-brown pigmentation progressing in a caudocranial fashion [Figure 1].[2] Lichen aureus is the least common type and is characterized by discrete and confluent macules or plaques that are golden, dark brown, or bronze colored.[2,5] Majocchi purpura has female preponderance whereas all other types have male preponderance [Table 1].[1]
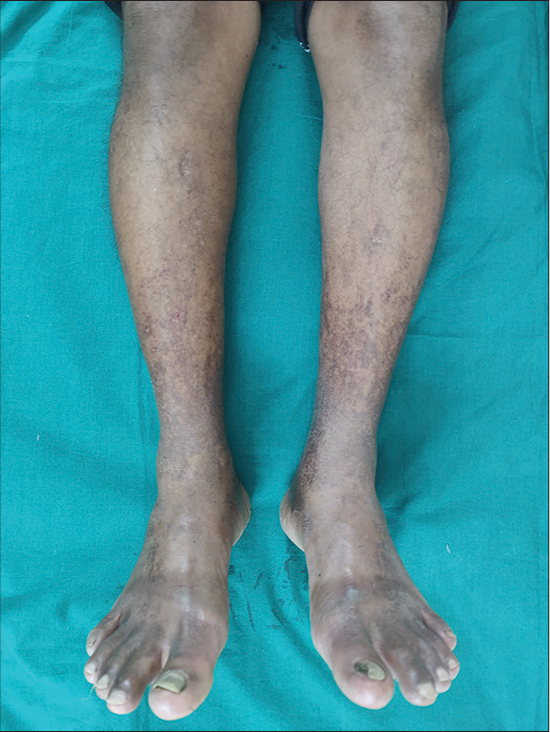
- Clinical picture of pigmented purpuric dermatosis.
| Variant | Clinical characteristics | Course |
|---|---|---|
| Schamberg disease | Asymptomatic, orange-red macules with pinpoint petechiae, called “cayenne pepper” spots, in older individuals in lower legs | Remissions and relapses |
| Pigmented purpuric lichenoid dermatosis of Gougerot and Blum | Itchy, purpuric lichenoid papules that merge into plaques, in elderly overlegs, resemble Kaposi sarcoma, mycosis fungoides, and cutaneous vasculitis. | Chronic |
| Eczematoid purpura of Doucas and Kapetanakis | Intensely itchy, lesions similar to Schamberg disease, papules and patches but with scaling in adult men, it tends to spread cranially from lower limb up to the abdomen and head. | Rapid onset and chronic course |
| Purpura annularis telangiectases (Majocchi disease) | In general, asymptomatic, erythematous annular macules with central fading and telangiectatic puncta at the periphery, over legs in children or young adults preferentially females | Chronic recurrent course |
| Lichen aureus | In general, asymptomatic, single golden-yellowish papules or plaques, sometimes segmental on the shin area of legs in children or young adults | Chronic |
AETIOPATHOGENESIS
The etiology is not clearly known. The proposed etiological factors include chronic venous hypertension, exercise, gravitational dependency, alcohol ingestion, capillary fragility, focal infections, and drugs such as aspirin, acetaminophen, adalin, hydralazine, persantin, thiamine reserpine, interferon alpha, glipizide, meprobamate, benzafibrate, and medroxyprogesterone acetate injection.[1,3,4] PPD can be associated with systemic diseases, such as diabetes mellitus, rheumatoid arthritis, systemic lupus erythematosus, thyroid dysfunction, mycosis fungoides, hematological and solid neoplasms, liver disease, and hyperlipidemia; however, most of the PPD remain idiopathic.[4]
Despite varying clinical features, all PPDs share similar histopathology.[5-7] The four characterizing elements are dilated blood vessels, with endothelial cell swelling, deposit of hemosiderin, red cell extravasation, and perivascular lymphocytic-macrophage infiltration. Epidermal spongiosis and lymphocytic exocytosis are common in all PPDs but are more pronounced in the eczematoid-like purpura of Doucas and Kapetanakis. Lichen aureus is characterized by a band-like dermal infiltrate separate from the normal epidermis by the Grenz zone. Lichenoid infiltrate and spongiosis with patchy parakeratosis are the distinguishing features of the dermatosis of Gougerot and Blum. A rare granulomatous form, a variant of a chronic pigmented purpura, has also been described.[5-7]
DERMOSCOPIC FEATURES
The dermoscopic features of PPD are well established. They include red dots and globules, coppery red pigmentation, brown patches, brown dots, red patches, linear vessels, annular/comma-like vessels, and lentigines-like reticular pigmentation.[6] The Figure 2 demonstrates a diagrammatic representation of the dermoscopic features of PPD. The Figures 3-5 depict various dermoscopic findings observed in PPD.
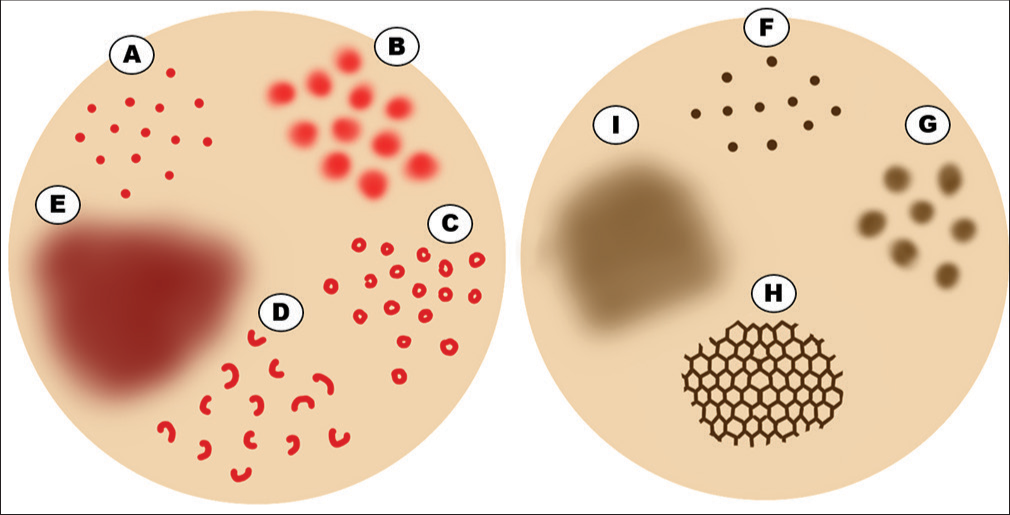
- Diagrammatic representation of dermoscopic features of pigmented purpuric dermatosis. (A) Red dots. (B) Red globules. (C) Red circles. (D) Comma-like vessels. (E) Coppery-red background. (F) Brown dots. (G) Brown globules. (H) Accentuated pigment network. (I) Brown background.
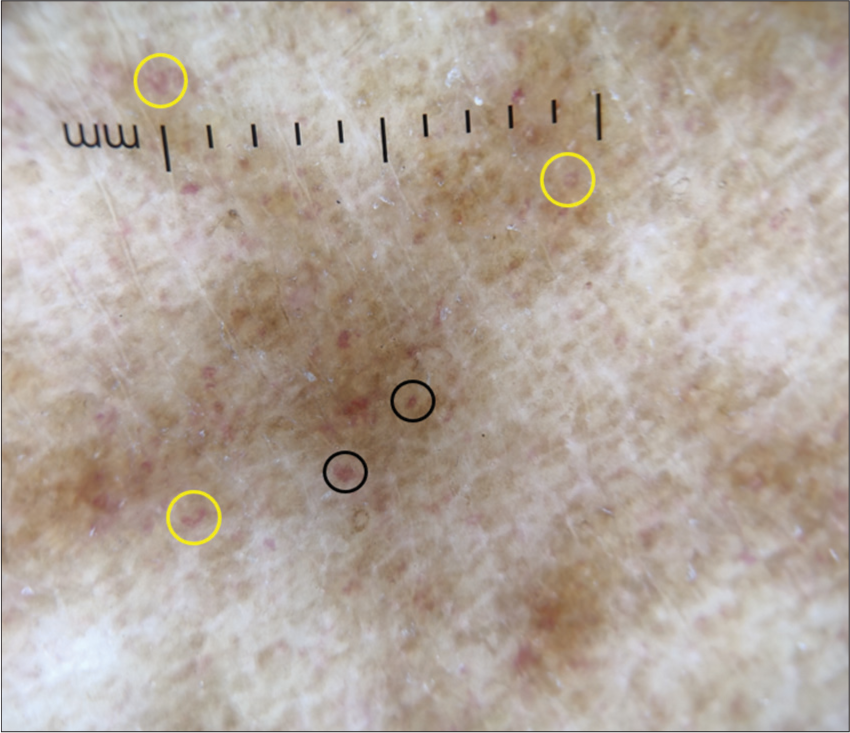
- Dermoscopy (DermLite DL4; ×10) shows red dots (black circles), comma-like vessels (yellow circles), brown background with increased pigment network.
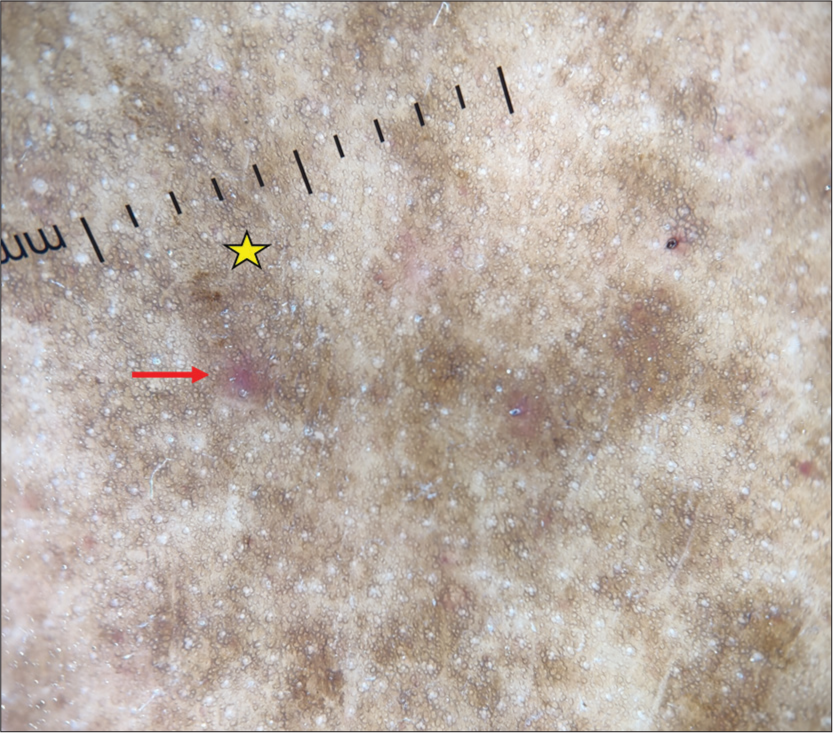
- Dermoscopy (DermLite DL4; ×10) shows red globules (red arrow), increased pigment network, and brown background (yellow star).
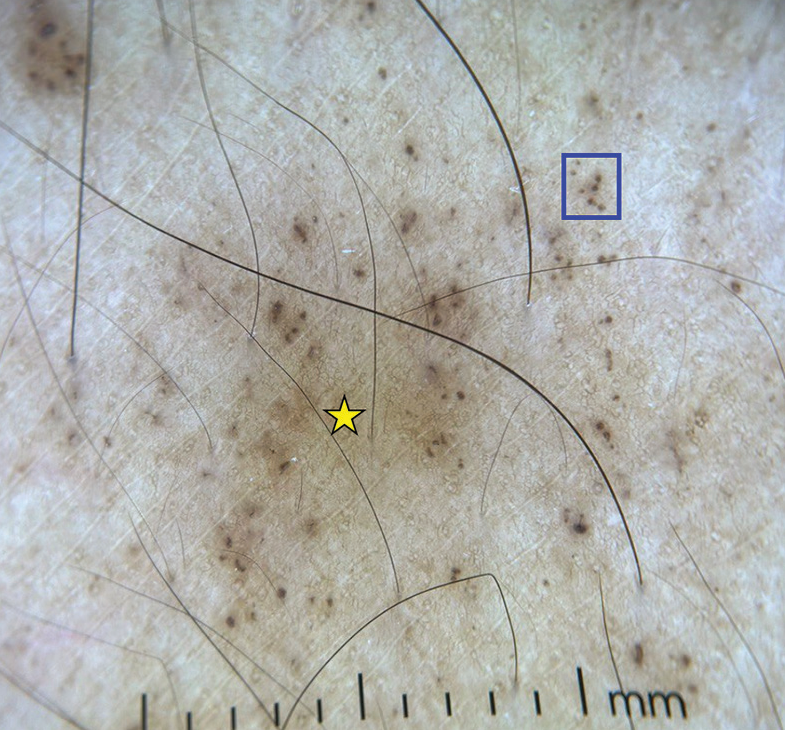
- Dermoscopy (DermLite DL4; ×10) shows brown dots (blue square) and brown background (yellow star).
DERMOSCOPIC AND HISTOPATHOLOGICAL CORRELATION
Main histopathological features include superficial perivascular lymphocytic and histiocytic infiltration with marked hemosiderin deposition and no leukocytoclastic vasculitis. Capillaritis is a feature of PPD.[7] Carlson and Chen said that PPD is cutaneous pseudovasculitis.[8] Extravasation of red blood cells (RBC) seen in histopathology is reflected by red dots and globules which is the most common pattern observed in dermoscopy of PPD.[2] Lymphohistiocytic infiltration in the dermis, RBC extravasation along with hemosiderin deposition are reflected by a coppery brown background in dermoscopy.[2,5] Increased dermal blood vessels observed correspond to a red background in dermoscopy.[2] Dilated coiled blood vessels in the upper dermis are reflected by red circles in dermoscopy.[2] The elliptical or spherical arrangement of pigmented melanocytes in the epidermal basal layer or the presence of melanophages in the upper dermis is seen as brown dots and partial pigmentation of rete ridges is seen as brown circles in dermoscopy.[2,4] Increased melanin in the epidermis and melanin pigment in the papillary dermis in free or nests reflected by brown reticular lines and gray dots, respectively.[2]
In lichen aureus, there are dense band-like lymphohistiocytic infiltrates in the upper dermis in the Grenz zone that can be confused with mycosis fungoides and less RBC extravasation as compared with other types of PPD.[5] Histopathology of granulomatous PPD shows superficial perivascular and focal band-like infiltrates consisting of lymphocytes and histiocytes. It also shows poorly formed non-necrotizing granulomas scattered through the dermis. The possibility of mycosis fungoides should be suspected due to its histopathology although it is not reported in the literature.[9]
Kim et al.[3] conducted a study on 38 patients with PPD. They included 20 females and 18 males. The mean age of the study was 42.6 ± 19.3 years. The most common sites observed were lower limbs (78.9%), followed by upper and lower limbs with trunk (5.3%) and upper limbs with trunk (2.6%). They included Schamberg disease (n = 23, 60.5%), Majocchi’s purpura (n = 8, 21.1%), PPD of Gougerot-Blum (n = 3, 7.9%), lichen aureus, and eczematoid-like purpura of Doucas-Kapetanakis (n = 2, 5.3) each. They also found the following histopathological findings in descending order: perivascular lymphocytes (79%), endothelial cell swelling (52%), extravasation of RBCs (50%), interface changes (42%), lichenoid lymphocytic infiltrates (39.5%), hyperpigmentation of basal layer (29%), deposition of hemosiderin pigment (26.3%), exocytosis of lymphocytes (18.4%), and spongiosis in epidermis (13.2%).[3]
Ozkaya et al.[4] studied 32 patients with PPD. The mean age of this study was 49.5. This study included 17 men (53%) and 15 women (47%). The mean disease duration was 12 months. The morphological types of PPD observed were Schamberg disease (91%; n = 29), Majocchi purpura (6%; n = 2), and linear type (3%; n = 1). Dermoscopy patterns observed were coppery-red background (97%), red globules (75%), red dots (69%), brown dots (53%), brown network (34%), red patches (34%), brown globules (26%), linear vessels (22%), linear brown lines (22%), and follicular openings (13%). They also observed elevated LDL (61%; n = 147), hypertriglyceridemia (43%; n = 10), and hypercholesterolemia (44%; n = 12). Thus, they proposed that hyperlipidemia is responsible for microvascular reactions and that this may have a role in the causality of PPD.[4]
Suh et al.[10] conducted a study to differentiate lichen aureus from nummular eczema with the help of dermoscopy. They enrolled 14 patients of lichen aureus and nummular eczema each. A dermoscopy of lichen aureus showed a diffuse orange background which histopathologically correlated with lymphohistiocytic dermal infiltrate, extravasation of RBC, and hemosiderin in macrophages. They also observed red globules, gray dots, and brown to gray interconnected lines due to increase in number of blood vessels, aggregation of hemosiderin-laden macrophages, and hyperpigmented basal layer and pigmentary incontinence in histopathology, respectively, whereas in nummular eczema, dermoscopy showed scales, irregular brownish-red globules, and shiny yellow clods. The latter correlated histopathologically with serum crust.[10]
Huang et al.[7] studied biopsy-proven PPD and its variants between January 2007 and December 2016. They included 107 patients (58 males and 49 females) and studied their histological patterns from previous medical records.[7] Four classical histopathological patterns were observed:
Spongiotic - which is not described very frequently had spongiosis and was associated with eczematoid-like purpura of Doucas and Kapetanakis, Schamberg disease
Lichenoid – dense band-like lymphocytic infiltrate in the upper dermis
Interface – is considered as a diagnostic pitfall and assumed as a secondary pathological change in PPD – has more basal vacuolar degeneration and dyskeratotic keratinocytes without much band-like inflammatory infiltrate.
Perivascular has perivascular inflammatory infiltrates.
The order of occurrence of histopathological patterns observed was lichenoid (42%), perivascular (37.40%), interface (10.30%), spongiotic (6.5%), and granulomatous (3.70%). A granulomatous variant of PPD is a rare clinical type. It was found in association with hyperlipidemia but was not statistically significant in this study.[7] Other common findings noted include papillary dermal fibrosis (85.0%); lymphocytic exocytosis (70.1%); erythrocyte exocytosis (56.1%); epidermal hyperkeratosis, parakeratosis, or acanthosis (70.1%); vacuolization of basal layer (43.0%); beneath the basal layer (71.0%); lymphocytic vasculitis (15.9%); and Langerhans cell microabscess (3.7%).[7] The Table 2 depicts the common features observed in each histological pattern found by Huang et al.[7]
| Study | Pattern | Features |
|---|---|---|
| Huang et al.[7] | Lichenoid pattern (n=45; 42%) | Papillary dermal fibrosis (100%) Lymphocytic vasculitis (24.4%) Features mimicking mycosis fungoides (11.1%) Dermal hemorrhage |
| Superficial perivascular pattern (n=40; 37.40%) | Papillary dermal fibrosis (67.5%) Epidermal change (47.5%) Spongiosis (45.0%) Dyskeratotic cells (20.0%) Hemorrhage (15.0%) |
|
| Interface pattern (n=11; 10.30%) | Exocytosis of lymphocytes (90.9%) Pigmentation (90.9%) Dyskeratotic cells Basal vacuolization |
|
| Spongiotic pattern (n=7; 6.5%) | Diffuse lymphocytic exocytosis with spongiosis Subepidermal vacuolization (100%) Papillary dermal fibrosis (100%) |
Metin and Elmas[2] conducted a study on dermoscopic patterns of PPD including 25 patients with 15 males (60%) and 10 females (40%). The mean age was 42 years, with a mean disease duration of 4 months. They observed Schamberg disease in 19 (76%) patients and lichen aureus in 6 (24%) patients. The dermoscopic patterns observed were red dots and red globules (100%), coppery brown pigmentation in the background (72%), and subtle brown dots and brown reticular lines (40% each). Other features observed in dermoscopy were brown circles (40%), red circles (32%), subtle gray dots (32%), and rosette structure (8%) which were not described previously.[2] The Table 3 shows a comparison of Metin and Elmas,[2] Ozkaya et al.,[4] and Çakmak et al.[11] studies.
| Findings in dermoscopy | Metin et al.[2] | Ozkaya et al.[4] | Çakmak et al.[11] |
|---|---|---|---|
| Red dots and globules | 100% | 69%, and 75% | 100% |
| Coppery brown background | 72% | 97% | 100% |
| Brown reticular lines | 40% | 34% | 44.4% |
| Brown dots | 40% | 53% | 16.6% |
| Subtle gray dots | 32% | – | 16.6% |
PPD: Pigmented purpuric dermatosis
Garrido et al.[12] reported a case of pigmented purpuric lichenoid dermatitis of Gourgerot and Blum in a human immunodeficiency virus (HIV) type 1 infection on highly active anti-retroviral treatment (HAART). They observed multiple red-brown irregular-shaped nummular plaques present over bilateral legs in HIV patients on HAART. Dermoscopy showed a coppery reddish brown background correlating with extravasation of RBC, hemosiderin deposition, and lichenoid infiltrate. Another finding in dermoscopy was a pearly white central area correlating with hemosiderin within macrophages seen in the dermis. They concluded that the PPD has developed as a result of immunological imbalance due to immune reconstitution inflammatory syndrome.[12]
DIAGNOSIS
Diagnosis relies primarily on clinical evaluation of skin lesions, with biopsy as a confirmatory tool.[13,14] Although the exact cause of PPD remains unclear, capillary fragility and RBC extravasation are implicated. The differential diagnosis for PPD includes several skin conditions that may share similar clinical or histopathological features, particularly involving the lower extremities. Most of the conditions are macular in nature, and others are papular, plaque, or nodular. Prominent entities include drug hypersensitivity reactions, due to carbamazepine or nitroglycerine. Purpuric contact dermatitis to clothing is characterized by lesions confined to areas in contact with clothing, accompanied by intense itching, often associated with materials such as wool and coloring agents. Venous stasis purpura manifests with signs of chronic venous insufficiency, including swelling, varicose veins, and venous ulcers. Other considerations encompass conditions such as purpura due to thrombocytopenia, senile purpura in the elderly, purpuric exanthema attributed to viral infections, leukocytoclastic vasculitis, immunoglobulin A vasculitis (Schönlein-Henoch purpura) commonly in pediatric cases, Kaposi sarcoma affecting elderly or immunosuppressed individuals, and purpuric mycosis fungoides.[13,14]
TREATMENT
Treatment strategies for PPD aim to alleviate symptoms, considering the generally benign and chronic nature of the condition.[13,14] Given that PPD is frequently asymptomatic and tends to have a self-limited nature in many cases, adopting close monitoring of the condition without immediately initiating active treatment could be considered. As there is no standardized treatment, various methods with varying efficacy are employed. Topical corticosteroids, commonly employed, yield unsatisfactory results, and the recurrence rate upon cessation of treatment remains a concern. Topical calcineurin inhibitors – pimecrolimus and tacrolimus – might be effective and have a better side effect profile than corticosteroids. Phototherapy, particularly in children, emerges as a viable option, demonstrating good tolerance and therapeutic outcomes, especially in extensive or unresponsive cases. Pentoxifylline, with its multifaceted mechanisms, has shown promise, although with higher dosage regimens of 1.2 g a day. The immunomodulatory and immunosuppressant agents, including colchicine (0.5 mg twice a day, high efficacy reported in few cases), dapsone, methotrexate, and cyclosporine, present encouraging results but should be reserved for highly symptomatic, refractory cases due to potential side effects. The role of hemorheological agents, such as vitamin C (500 mg twice or thrice daily), calcium dobesilate (500 mg/day), and flavonoids, remains noteworthy, providing an alternative avenue for consideration. Another promising option in PPD treatment is laser therapy. Photodynamic therapy was explored in two females exhibiting a localized variant of PPD on their lower legs. The precise workings of lasers in PPD are yet to be completely understood. Nevertheless, potential explanations include the elimination of hemosiderin deposits, collagen remodeling, and anti-inflammatory effects. Laser therapy may be a promising choice for localized lesions resistant to topical treatments. Associated venous stasis should be treated by compression, and prolonged leg dependency should be avoided. Watchful waiting might be an acceptable approach in asymptomatic cases, acknowledging the self-limiting nature of the disease.[13,14]
PROGNOSIS
Although in the majority of cases, they are idiopathic, routine blood tests and coagulation tests are recommended to exclude a possible association with thrombocytopenia or blood clotting disorder.[13,14] As far as the treatment, most cases are resistant and usually relapse thus non-pharmacological measures must be always considered.[13,14]
CONCLUSION
PPD is an acquired chronic relapsing skin condition. It is characterized by asymptomatic or itchy symmetric petechiae, macules, and red-brown patchy pigmentation. This article presents a dermoscopic perspective of PPD and reviews the current literature related to this entity. The management of Schamberg disease and other PPDs poses a therapeutic challenge, due to the restricted efficacy of existing interventions, emphasizing the necessity for a combined therapeutic approach.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Dermoscopic findings and the clinicopathologic correlation of pigmented purpuric dermatosis: A retrospective review of 60 cases. Ann Dermatol. 2021;33:214-21.
- [CrossRef] [Google Scholar]
- Dermoscopic profile of pigmented purpuric dermatosis: New observations. Postepy Dermatol Alergol. 2019;36:687-91.
- [CrossRef] [Google Scholar]
- Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-10.
- [CrossRef] [Google Scholar]
- Dermatoscopic findings of pigmented purpuric dermatosis. An Bras Dermatol. 2016;91:584-7.
- [CrossRef] [Google Scholar]
- Dermoscopy as an evolving tool to assess vitiligo activity. J Am Acad Dermatol. 2018;78:1017-9.
- [CrossRef] [Google Scholar]
- The pathological spectrum and clinical correlation of pigmented purpuric dermatosis-A retrospective review of 107 cases. J Cutan Pathol. 2018;45:325-32.
- [CrossRef] [Google Scholar]
- Granulomatous pigmented purpuric dermatosis: Report of a case with atypical clinical presentation including dermoscopic findings. Am J Dermatopathol. 2015;37:311-4.
- [CrossRef] [Google Scholar]
- Diagnostic usefulness of dermoscopy in differentiating lichen aureus from nummular eczema. J Dermatol. 2017;44:533-7.
- [CrossRef] [Google Scholar]
- Dermoscopic findings in patients with pigmented purpuric dermatoses. Acta Dermatovenerol Croat. 2016;24:291-5.
- [Google Scholar]
- Dermoscopy of pigmented purpuric lichenoid dermatitis of gougerot and blum in an HIV-infected patient. Dermatol Pract Concept. 2020;10:e2020075.
- [CrossRef] [Google Scholar]
- Therapeutic approach in pigmented purpuric dermatoses-a scoping review. Int J Mol Sci. 2024;25:2644.
- [CrossRef] [Google Scholar]
- Pigmented purpuric dermatoses: A complete narrative review. J Clin Med. 2021;2283
- [CrossRef] [Google Scholar]




