
Translate this page into:
Dyschromatosis universalis hereditaria (DUH)

*Corresponding author: Anmol Batra, Department of Dermatology, AIIMS Rishikesh, Uttarakhand, India. anmolbatra9@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Batra A, Hazarika N. Dyschromatosis universalis hereditaria (DUH). CosmoDerma 2022;2:38.
A 41-year-old male presented to us with multiple asymptomatic hypopigmented, and hyperpigmented skin lesions all over the body since the age of 5 years. The lesions had started over the abdomen and gradually spread toward other body parts over the duration of the next 3 years. There was no history of photosensitivity or any drug intake or handling of any chemical. No one in the family had similar complaints. On cutaneous examination, there was diffuse hyperpigmentation interspersed with spotty and reticulate hypopigmented macules (giving a mottled appearance) of irregular size and shape over the trunk and limbs relatively sparing bilateral axilla and inguinal folds [Figure 1a-e]. Keratotic follicular papules, suggestive of keratosis pilaris [Figure 1c-e], were also seen on the posteromedial aspects of the arms and anterior aspects of the thighs. There were concomitant unrelated tinea corporis plaques [Figure 1e] on the medial aspect of the thighs as well. His palms, soles, and mucous membranes were within normal limits. Based on these findings, a diagnosis of dyschromatosis universalis hereditaria (DUH) was made. The patient was counseled regarding the unremitting, but benign nature of DUH.
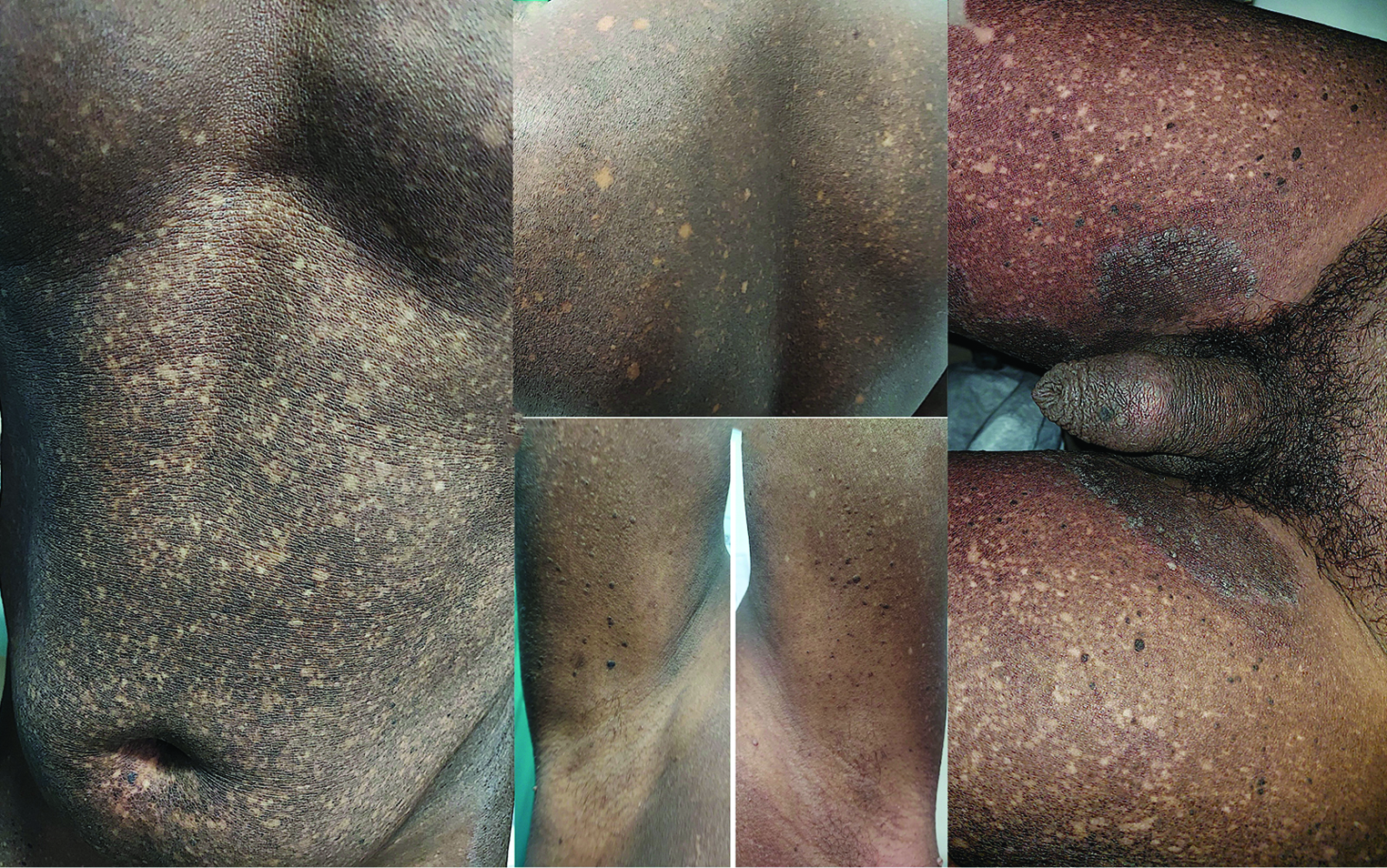
- (a and b) Dyschromasia—diffuse and spotty hyperpigmentation interspersed with spotty and reticulate hypopigmented macules of irregular size and shape over the trunk (c and d) limbs relatively sparing bilateral axilla (e) inguinal folds.
Dyschromatosis universalis hereditaria is a rare autosomal genodermatosis. It was first described in 1929 in Japan. It is classified into three subtypes. DUH1-an autosomal dominant disease with linkage to 6q24.2-q25.2.[1] DUH2-autosomal recessive disease mapped to 12q21- q23.[2] DUH3-heterozygous mutation in the ABCB6 gene on chromosome 2q35.[3] DUH has been associated with neurological, ophthalmological, and hematological complications.[2]
Declaration of patient consent
Patient’s consent is not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 2003;73:693-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Dyschromatosis universalis hereditaria: evidence for autosomal recessive inheritance and identification of a new locus on chromosome 12q21-q23. Clin Genet. 2008;73:566-72.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J Invest Dermatol. 2013;133:2221-8.
- [CrossRef] [PubMed] [Google Scholar]




