
Translate this page into:
A curious case of scarring and non-scarring alopecia in the same patient: Graham Little-Piccardi-Lassueur syndrome

-
Received: ,
Accepted: ,
How to cite this article: Narayanan A, Srinivas BH, Ram Kumar KR, Tirekha SV, Thappa DM. A curious case of scarring and non-scarring alopecia in the same patient: Graham Little-Piccardi-Lassueur syndrome. CosmoDerma 2022;2:57.
Dear Sir,
Graham Little-Piccardi-Lassueur syndrome (GLPLS) is a rare variant of lichen planopilaris presenting with patchy cicatricial alopecia of the scalp, non-scarring alopecia of the axilla and/or groin, and follicular keratotic papules over the body. We report a rare case of GLPLS in a male patient with concomitant lichen planus on his nose.
A 25-year-old male patient presented with multiple pruritic patches of cicatricial alopecia over the vertex [Figure 1a] and temporal scalp, which were progressively increasing in size, for the past 1 year. He did not have any history of photosensitivity or pus discharge from the alopecia patches. The patient had not received any treatment earlier for this condition. On examination, we observed patches of non-scarring alopecia over the pubic area [Figure 1b] and multiple, small, follicular keratotic papules (1 mm in size) over his upper back. In addition, he had a well-defined, violaceous macule of size 3 × 1 cm over the ala of the left nose. Dermoscopy (Dinolite Pro, non-contact, non-polarized, ×100) revealed a loss of eccrine openings in the scalp lesion [Figure 1c]. In addition, dermoscopy (DermLite DL200 hybrid, non-contact, ×10) of the active border of alopecia revealed perifollicular scales [red arrow, Figure 1d]. Histopathology of the scalp lesion revealed perifollicular patchy mild-to-moderate lymphocytic infiltrate with fibrosis [Figure 2a and b]. These histopathological features were consistent with lichen planopilaris. Histopathology of the nose lesion revealed hypergranulosis with basal vacuolar degeneration and few necrotic keratinocytes in the epidermis [Figure 2c]. Superficial dermis showed patchy moderate to dense lymphohistiocytic infiltrate [Figure 2d]. These features were consistent with lichen planus. Based on the characteristic presentation and histopathological findings, we made a final diagnosis of GLPLS with cutaneous lichen planus. The patient was started on oral steroids and hydroxychloroquine.
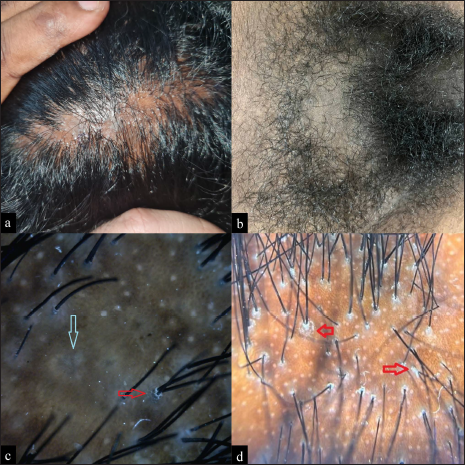
- (a) Pruritic patches of cicatricial alopecia over the vertex with (b) nonscarring alopecia over the pubic area. Dermoscopy (DinoLite Pro, noncontact, nonpolarized, ×100) of the cicatrical alopecia showed loss of eccrine openings, blue-gray target pattern (blue arrow) and perifollciular scales (red arrow). In addition, dermoscopy (DermLite DL200 hybrid, noncontact, ×10) of the active border of alopecia revealed perifollicular scales (red arrow).
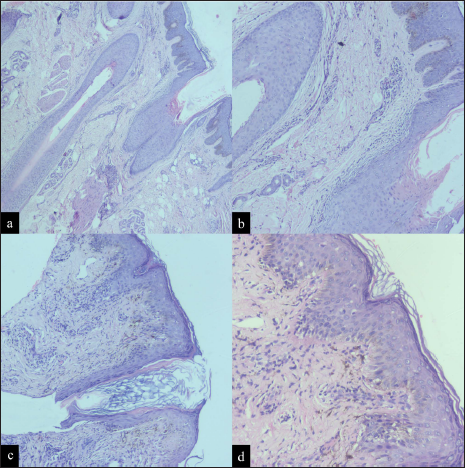
- Histopathology of the scalp lesion revealed (a) orthokeratotic epidermis (H&E, ×40) with (b) perifollicular lymphocytic infiltrate (H&E, ×100). Histopathology of the nose lesion revealed (c) irregular acanthosis in the epidermis (H&E, ×100) with (d) hypergranulosis, basal vacuolar degeneration and few necrotic keratinocytes (H&E, ×200). Superficial dermis showed patchy moderate to dense lymphohistiocytic infiltrate.
Graham Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913. It was later described by Graham-Little in 1915, in a patient referred by Lassuer. This gives the condition its name.[1] The condition is seen in middle-aged females. The presentation of GLPLS in males, as seen in our case, is rare. The underlying pathogenesis is hypothesized to be T-cell-mediated autoimmunity. Studies have shown decreased expression of peroxisome proliferator-activated receptor. Interferon and Janus kinase (JAK) signaling are also known to be upregulated in lichen planopilaris.
Graham Little-Picardi-Lassueur syndrome is characterized by progressive cicatricial alopecia of the scalp and alopecia of pubic and axillary hair without clinically evident scarring. Patients also present with keratosis pilaris. It has been shown that more than 50% of patients with GLPLS experience one episode of mucosal or cutaneous lichen planus in their lifetime.[2] However, the presence of concomitant cutaneous lichen planus during presentation is uncommon.[3] GLPLS has been reported to occur with concomitant hypertrophic lichen planus and lichen planus pigmentosus.
Dermoscopy of lichen planopilaris reveals perifollicular scales which correspond to infundibular hyperplasia. Scattered brown-black interfollicular pigmentation, dark perihilar halo, and a blue-gray target pattern can also be seen.[4] Histology of lichen planopilaris reveals an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle. There is sparing of the hair bulb. The pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and cause its ultimate death.[5] Histology of older lesions reveals fibrotic longitudinal tracks, perifollicular lamellar fibrosis, and epidermal atrophy.
Early initiation of treatment is required to prevent the progression of hair loss. Treatment options utilized in GLPLS include topical corticosteroids, intralesional corticosteroids, hydroxychloroquine, cyclosporine, pioglitazone, and tofacitinib.[6] The follicular keratotic paples can be treated with topical tretinoin 0.05%. Chiang et al. demonstrated the efficacy of hydroxychloroquine in 40 patients with lichen plano pilaris and found 69% and 83% reduction in the severity of lichen planopilaris at 6 and 12 months.[7]
Graham Little-Piccardi-Lassueur syndrome is a rare variant of lichen planopilaris. Dermatologists should be aware of this entity to make an early diagnosis and initiate appropriate treatment in such patients.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflict of interest
Devinder Mohan Thappa is the Editor-in-Chief of this journal.
References
- Graham-Little Piccardi Lassueur syndrome and review of the literature. Clin Case Rep. 2021;9:e04761.
- [CrossRef] [PubMed] [Google Scholar]
- Antigen mimicry followed by epitope spreading: A pathogenetic trigger for the clinical morphology of lichen planus and its transition to Graham Lassueur Piccardi Little Syndrome and keratosis lichenoides chronica - Medical hypotheses or reality? An Bras Dermatol. 2009;84:682-8.
- [CrossRef] [PubMed] [Google Scholar]
- Graham-Little-Piccardi-Lassueur syndrome with concomitant mucocutaneous lichen planus: Rare presentation in a man. Dermatol Pract Concept. 2022;12:e2022042.
- [CrossRef] [PubMed] [Google Scholar]
- Graham-Little-Piccardi-Lassueur syndrome in siblings. Int J Dermatol. 2021;60:e347-9.
- [CrossRef] [PubMed] [Google Scholar]
- Graham-Little Piccardi Lassueur syndrome: An unusual variant of follicular lichen planus. Int J Trichol. 2011;3:28-30.
- [CrossRef] [PubMed] [Google Scholar]
- Hydroxychloroquine and lichen planopilaris: Efficacy and introduction of Lichen Planopilaris Activity Index scoring system. J Am Acad Dermatol. 2010;62:387-92.
- [CrossRef] [PubMed] [Google Scholar]




