
Translate this page into:
Barraquer–Simons syndrome: A rare case of acquired partial lipodystrophy after pregnancy with diabetes mellitus

*Corresponding author: Dr. Surender Singh, Assistant Professor, Department of Dermatology, ESIC Medical College Sanathnagar, Hyderabad, Telangana, India. surender1994singh@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Gautam A, Badabagni P, Singh S, Mavoori A, Katike P, Himasribhamidipati S. Barraquer–Simons syndrome: A rare case of acquired partial lipodystrophy after pregnancy with diabetes mellitus. CosmoDerma. 2024;4:136. doi: 10.25259/CSDM_150_2024
Abstract
Barraquer–Simons syndrome (BSS) is an uncommon acquired partial lipodystrophy (APL) characterized by gradual, progressive loss of subcutaneous fat limited to the upper part of the body. It is a rare condition of uncertain origin typically appearing in early adolescence with a higher incidence among females with autoimmunity and C3 hypocomplementemia. Approximately 250 cases were reported worldwide, we present a case of BSS that began after pregnancy. A biopsy of the affected areas reveals reduced adipocytes. Metabolic abnormalities are relatively uncommon in APL compared to other forms of lipodystrophy, affecting around 10% of patients. At present, there is no treatment available to stop the disease’s progression. Management options are limited to restoration through lipomodelling or fat grafting in the affected regions. This report focuses on a 58-year-old patient with BSS who started post-pregnancy, and who also has diabetes mellitus and low C3 levels.
Keywords
Barraquer–Simons syndrome
Acquired partial lipodystrophy
Complement levels

INTRODUCTION
Barraquer–Simons syndrome (BSS) is an uncommon acquired partial lipodystrophy (APL) characterized by gradual, progressive loss of subcutaneous fat limited to the upper part of the body. It is a rare condition of uncertain origin typically appearing in early adolescence with a higher incidence among females with autoimmunity and C3 hypocomplementemia. Approximately 250 cases were reported worldwide.[1] We hereby report a case of a 58-year-old patient with BSS started after pregnancy with diabetes mellitus and low C3 levels.[2]
CASE REPORT
A 58-year-old female patient presented with loss of fat over the face, trunk upper extremities, and breast for 40 years. The patient was apparently well 40 years back when she developed swelling all over the body during her first pregnancy. After two months of normal delivery, she started losing subcutaneous fat initially over the face (temporal > orbital > buccal) which gradually progressed to involve the upper extremities and breasts over a period of 40 years.
She was a known case of diabetes mellitus on medication. There was no history of any drug intake. There is no history of fever, joint pains, or frothy urine. No history is suggestive of photosensitivity history of difficulty in getting up from a squatting position. There was no history of morning stiffness.
Cutaneous examination revealed facial lipoatrophy characterized by the loss of buccal fat pads with prominent zygomatic arches [Figures 1 and 2]. Bilateral breast hypoplasia [Figure 3] and abnormal fat deposition were noted over the shoulders and scapular region [Figure 4]. Subcutaneous fat in other areas such as the lower abdomen and thighs was normal [Figure 5]. The thyroid gland was normal on palpation. Hepatosplenomegaly, umbilical hernia, acanthosis nigricans, clitoromegaly, hirsutism, and acromegalic features were all absent. Ophthalmic and other systemic examinations were unremarkable, and her neurological examination and hearing status were confirmed as normal.
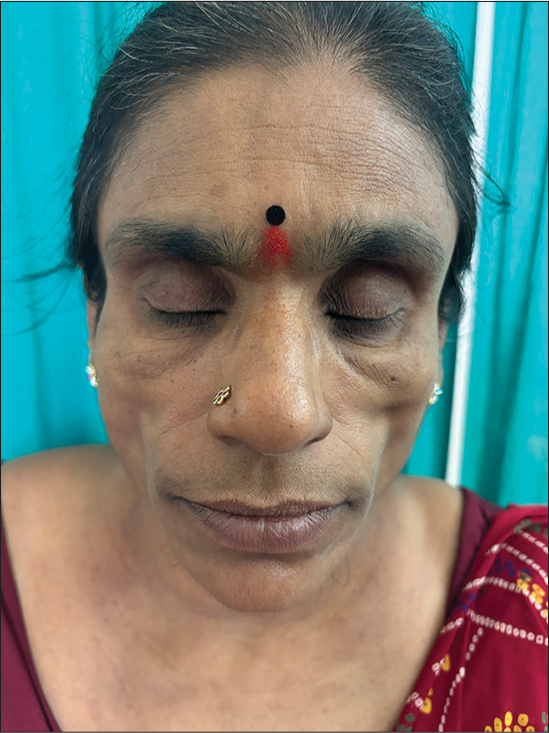
- Fat atrophy over buccal, orbital, and temporal area.

- Fat atrophy over the left upper limb.
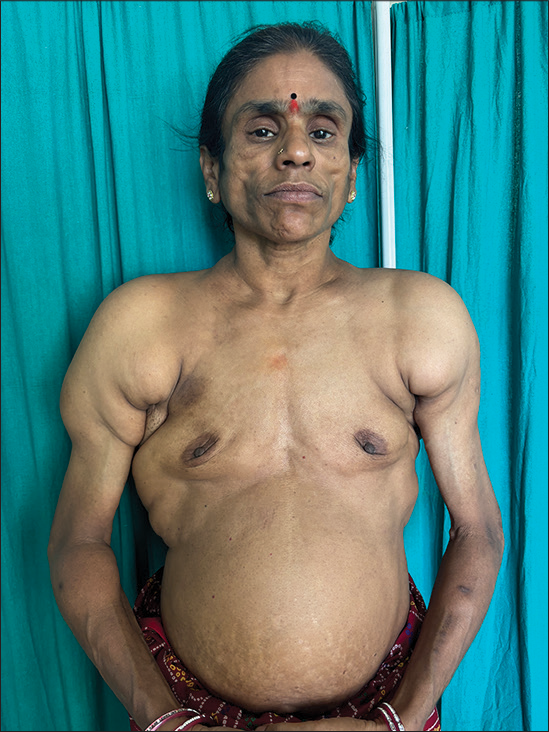
- Bilateral breast hypoplasia with abnormal deposition of fat over the shoulder.
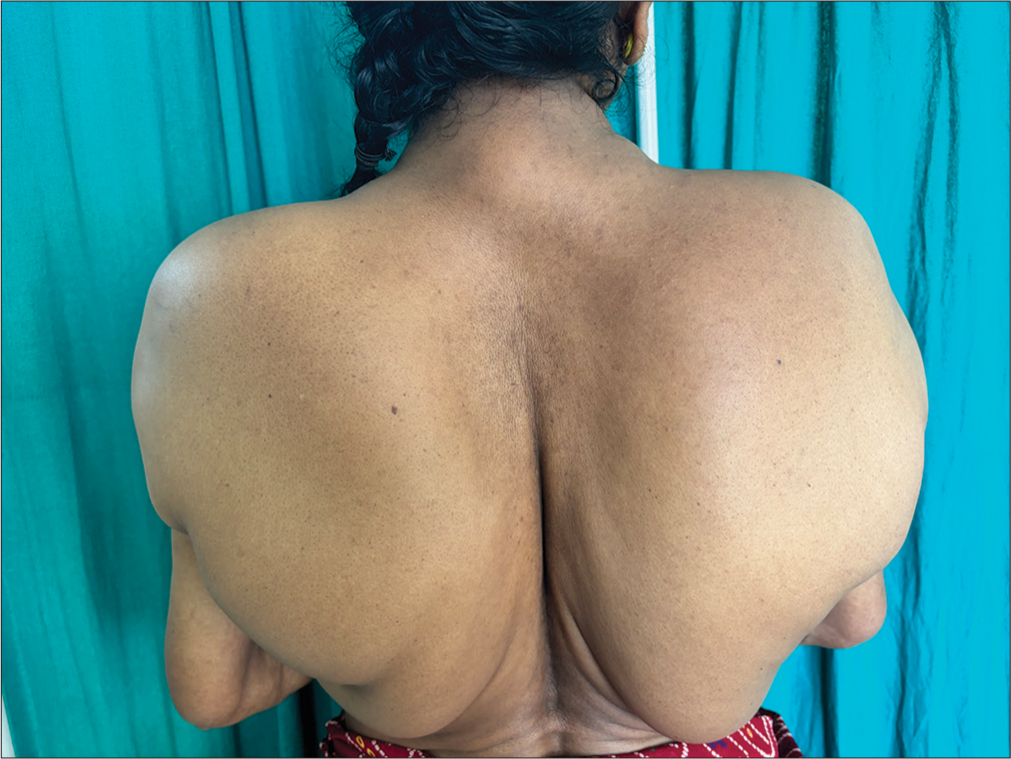
- Abnormal deposition of fat over the scapula.
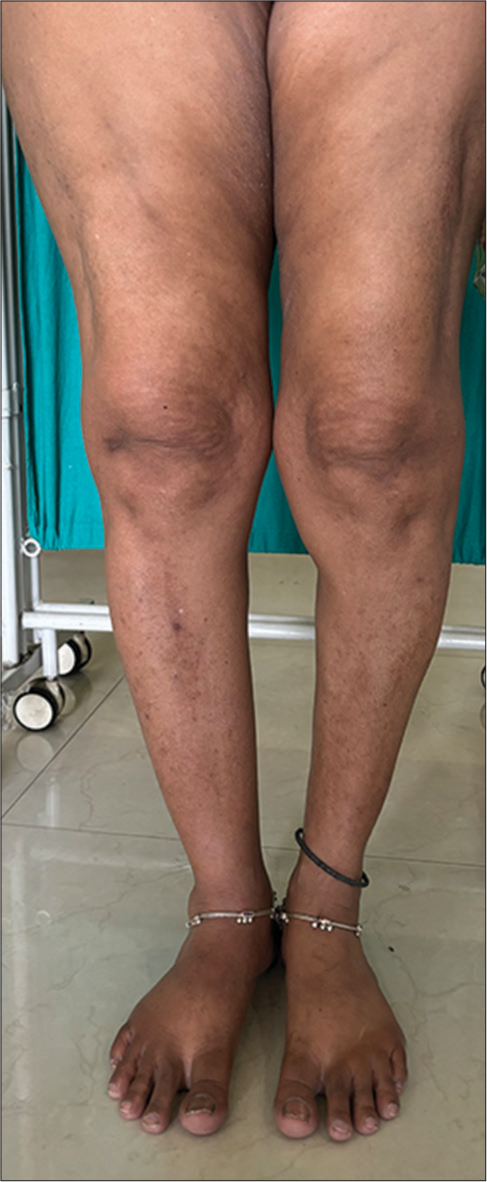
- Normal distribution of fat over bilateral lower limb.
Laboratory tests, including a complete blood count, biochemical parameters (such as renal and liver function tests), and urine analysis with urinary albumin excretion, showed no abnormalities. Her triglyceride levels were 155 mg/dL and serum high-density lipoprotein 48 mg/dL. Her thyroid function test was within normal limits. Her fasting blood glucose was 189 mg/dL, and her hemoglobin A1c was 10.9% showing uncontrolled diabetes. The rheumatoid factor was positive at 10 IU/mL. Complement C3 levels were found to be low, measured at <20 mg/dL (normal range: 83–177 mg/dL). C4 levels were normal. The patient showed no signs of renal disease. Skin biopsy was taken, from right forearm (atrophic site) and left shoulder (dystrophic site). The histopathological analysis of the atrophic region revealed mild orthokeratosis, thinning of the epidermis accompanied by vacuolar degeneration in the basal layer, and a reduction in adipocyte lobules within the subcutaneous layer [Figure 6].
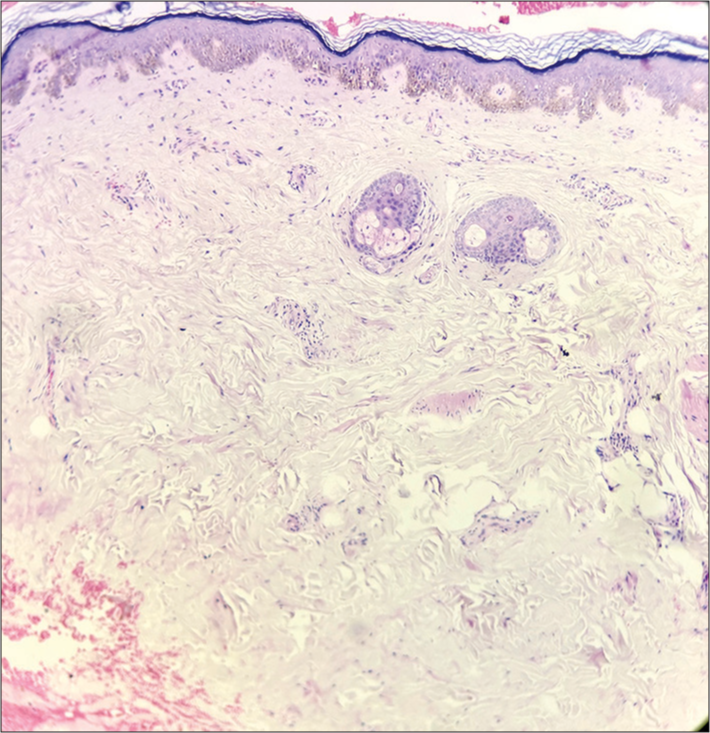
- Histopathological analysis (hematoxylin and eosin ×10) revealed mild orthokeratosis, thinning of the epidermis accompanied by vacuolar degeneration in the basal layer, and a reduction in adipocyte lobules within the subcutaneous layer.
DISCUSSION
Our case demonstrates a typical distribution of subcutaneous fat involvement with evidence of hypocomplementemia. Our case is noteworthy because disease progression started after the pregnancy and to the best of our knowledge, only one case is reported in the literature. The patient’s clinical and biochemical characteristics led us to identify the case as BSS as the primary diagnosis. The atypical distribution of fat aligned with the key criterion outlined by Misra et al.[3] which involves gradual, symmetrical loss of subcutaneous fat from the face, neck, upper arms, chest, and abdomen, while the lower extremities remain unaffected. Other criteria were also fulfilled, including the onset during adolescence, the absence of a family history of lipodystrophy, and low serum levels of C3. The low C3 levels help clearly differentiate this syndrome from other types of lipodystrophy. Our patient is having low C3 levels as well as other metabolic disorders like diabetes which is unusual. The C3-nephritic factor in serum triggers the destruction of adipocytes that express factor D (adipsin) – a serine protease enzyme – which leads to the widespread expression of factor D in various tissues, resulting in the distinctive pattern of fat loss.[4,5]
Patients with inherited lipodystrophies have been found to have mutations in several genes, such as LMNA, PPARG, AKT2, and ZMPSTE24 in cases of partial lipodystrophy[6] and AGPAT2, BSCL2, CAV1, and PTRF in cases of congenital total lipodystrophy.[7-9] However, the precise molecular mechanisms underlying APL remain unclear.
APL functions as a complex trait, where a susceptibility allele needs additional factors to activate the disease.[10] Typically, there is no family history of the condition, and it is commonly linked with a range of autoimmune disorders, including membranoproliferative glomerulonephritis, hypocomplementemia, systemic lupus erythematosus, dermatomyositis, and localized scleroderma.[2] Occasionally, the condition may also be associated with functional anomalies such as deafness, epilepsy, and intellectual disability.
Treatment for APL involves enhancing esthetic appearance through plastic surgery and managing associated systemic disorders. The primary aim of cosmetic surgical interventions is to alleviate the psychological distress that negatively impacts the patient’s quality of life. As per the literature, patients who have received treatment over a time period of nearly three years have experienced long-lasting positive effects from the treatment. Similar findings have been presented in a previously published case report of autologous fat graft treatment for BSS with stable results at two-year follow-up but near disappearance of volume after four years. The prognosis of BSS is mainly dependent on renal disease. A few patients have required renal transplantation for end-stage renal disease related to glomerulonephritis.[5,11]
CONCLUSION
BSS syndrome is an extremely rare form of acquired lipodystrophy with clinical and psychological consequences. Our case highlights the onset of disease after delivery as BSS is hypothesized to be associated with autoimmune disease. We conclude that pregnancy can be a trigger factor for the onset of BSS. Early diagnosis and regular follow-up are important to see for end-stage renal disease progression.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Clinical review#: Lipodystrophies: Genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96:3313-25.
- [CrossRef] [PubMed] [Google Scholar]
- Barraquer-Simons syndrome occurred after pregnancy: A rare case report. Fiziksel Tip Rehabil Bilim Derg. 2019;22:33-5.
- [CrossRef] [Google Scholar]
- Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: Report of 35 cases and review of the literature. Medicine (Baltimore). 2004;83:18-34.
- [CrossRef] [PubMed] [Google Scholar]
- Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies. J Hum Genet. 2013;59:16-23.
- [CrossRef] [PubMed] [Google Scholar]
- Complement mediated adipocyte lysis by nephritic factor sera. J Exp Med. 1993;177:1827-31.
- [CrossRef] [PubMed] [Google Scholar]
- Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93:1129-34.
- [CrossRef] [PubMed] [Google Scholar]
- Human PTRF mutations cause secondary deficiency of caveolin resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119:2623-33.
- [CrossRef] [PubMed] [Google Scholar]
- Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations. Clin Genet. 2010;78:66-73.
- [CrossRef] [PubMed] [Google Scholar]
- Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Human Genet. 2006;79:383-9.
- [CrossRef] [PubMed] [Google Scholar]
- Barraquer-Simons syndrome: A rare clinical entity. Am J Med Genet Part A. 2014;164:1756-60.
- [CrossRef] [PubMed] [Google Scholar]




